Диагностика гистиоцитоза из клеток Лангерганса у мальчика трех лет
С.В. Стоногин, А.И. Царева, А.К. Горюнов, Е.А. Таширова
ДГКБ имени З.А. Башляевой, Москва.
Мальчик трех лет поступил в хирургическое отделение по экстренным показаниям. В течение месяца отмечались боль, объёмное образование в левой заушной области, увеличение в размерах образования в течение 7 суток.
В левой заушной области пальпируется подкожное образование диаметром около 2.5см, мягко-эластичной консистенции, умеренно болезненное при пальпации. Отек и гиперемия диаметром до 2 см. Симптом флюктуации положительный.
Мальчик был оперирован по экстренным показаниям. Разрез над местом флюктуации проведён разрез 0.5 см. скудное гноевидное отделяемое. Выявлено белое опухолевидное образование (атерома?). При посеве отделяемого из раны роста микрофлоры не выявлено.
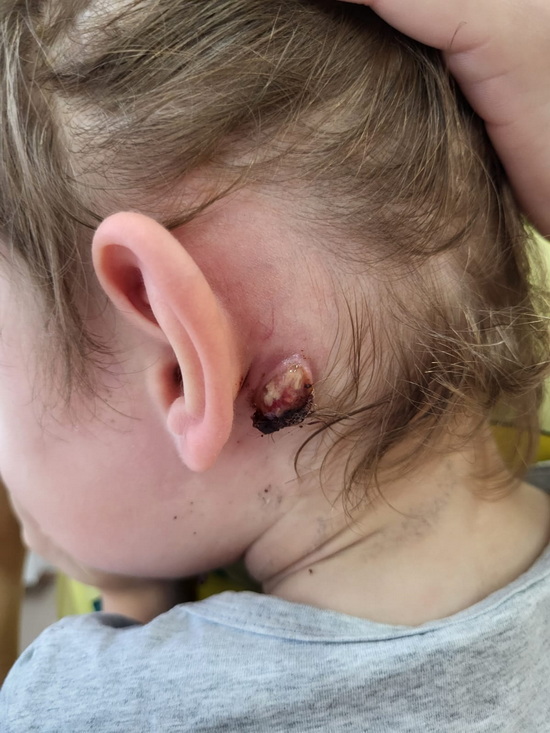
Рис 1 – вида раны в левой заушной области при перевязке.
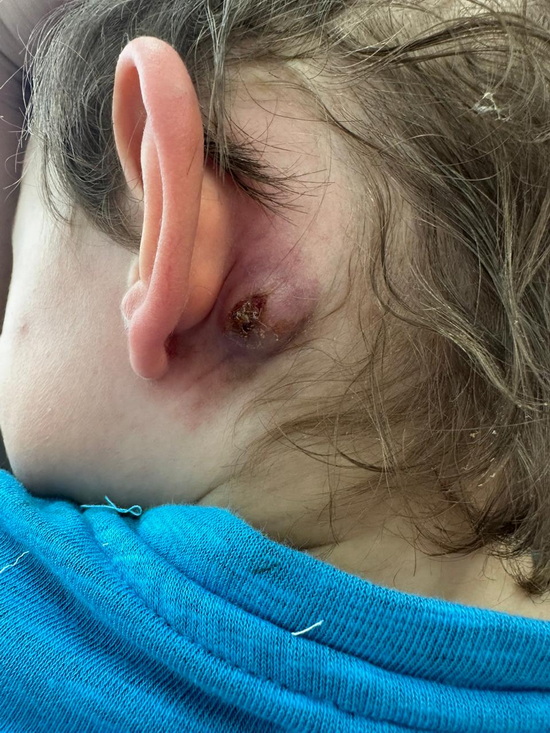
Рис 1а – вида раны в левой заушной области при перевязке.
Оториноларингологический статус. Форма наружного носа и области проекции ОНП не изменена, при пальпации безболезненная. Слизистая розовая, не отечная. В носовых ходах слизистое отделяемое скудно. Носовое дыхание удовлетворительное. Заушные области справа не изменены. В заушной области слева рана, из раны выбухает эластичное образование покрытое фибрином диаметром 2 на 2 см. Кожа сосцевидного отростка гиперемирована. Слуховые проходы широкие, свободный. Барабанные перепонки тусклая.
Ротоглотка: Зев симметричен, миндалины 2 степени, гиперемирован. Задняя стенка глотки без особенностей.
Учитывая нехарактерный для абсцесса вид раны, отсутствие эффекта от антибактериальной терапии, перевязок с антисептиками, сохранение боли при пальпации данной области, было решено выполнить спиральную компьютерную томографию головы.
На серии томограмм толщиной среза 1 мм получены изображения головного мозга. Срединные структуры не смещены. Боковые желудочки мозга симметричны, не расширены, третий не расширен, четвёртый не деформирован.
Цистерны основания не расширены, не деформированы. Обводные цистерны не расширены, симметричны. Щели мозга: латеральные не расширены, симметричные, межполушарная не расширена. Конвекситальные борозды не изменены. Дифференцировка вещества мозга на серое и белое вещество сохранена. Дополнительных объёмных структур и очагов патологической плотности не выявлено. Просвет подоболочечных пространств не расширен. Кости свода черепа не изменены. Придаточные пазухи и ячейки решетчатой кости, а так же ячейки сосцевидного отростка справа и полости среднего уха с обеих сторон пневматизированы. Структуры органов пирамид височных костей сохранены, симметричны.
В проекции левого сосцевидного отростка и заушной области отмечается участок деструкции кости с разрушением наружной костной пластинки, стенок ячеек сосцевидного отростка и внутренней костной пластинки в проекции лоха сигмовидного синуса. В просвете лизированной кости отмечается мягкотканная структура неоднородной плотности в среднем около 45 HU.
КТ картина объемного образования в проекции сосцевидного отростка слева
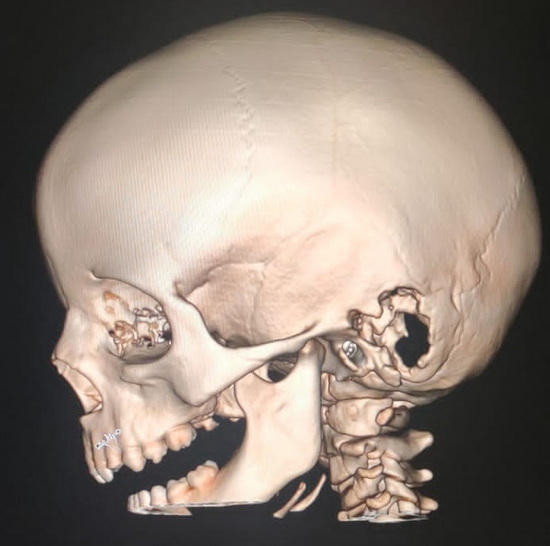
Рис 2 – компьютерная томография головы. 3Д реконструкция
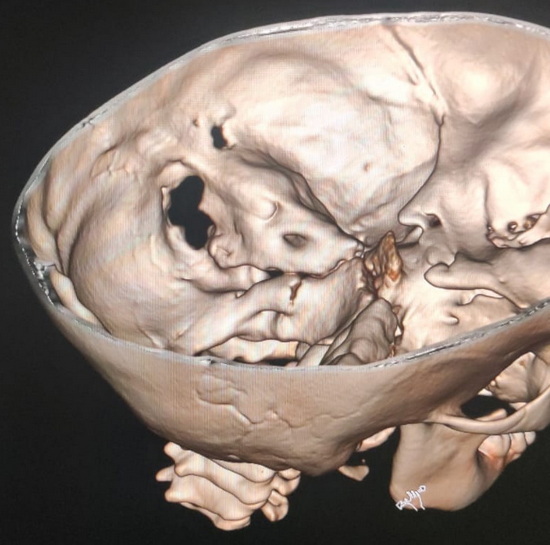
Рис 3 – компьютерная томография головы. 3Д реконструкция
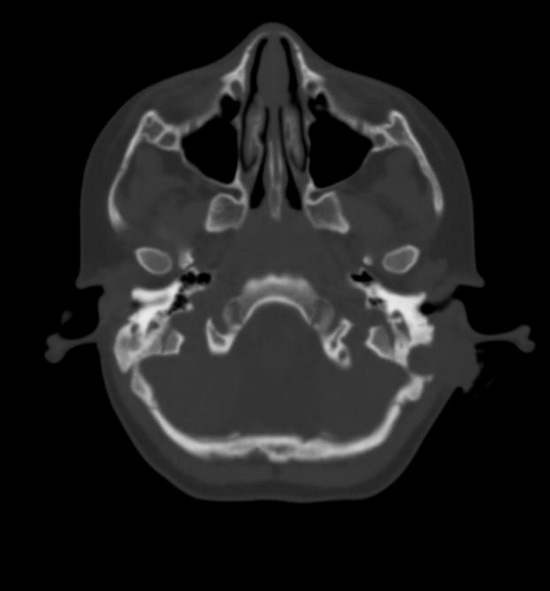
Рис 4 - компьютерная томография головы.
После проведения СКТ головы было решено выполнить биопсию данного образования, которое было целиком удалено и отправлено на гистологическое исследование.
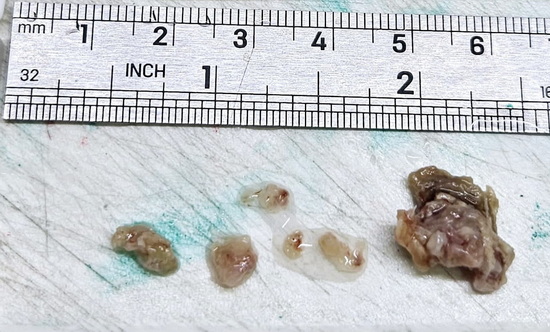
Рис 5 – макропрепарат удаленного образования
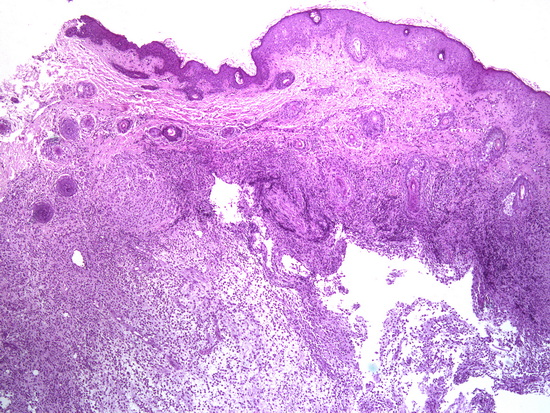
Рис 6- увеличение х40, гематоксилин-эозин кожа, в дерме опухоль.
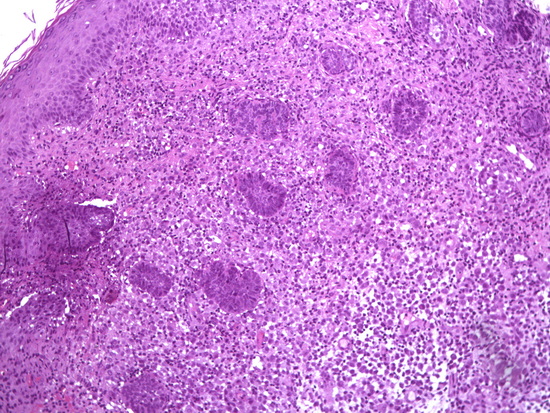
Рис 7 - увеличение х100, гематоксилин-эозин кожа, в дерме опухоль.
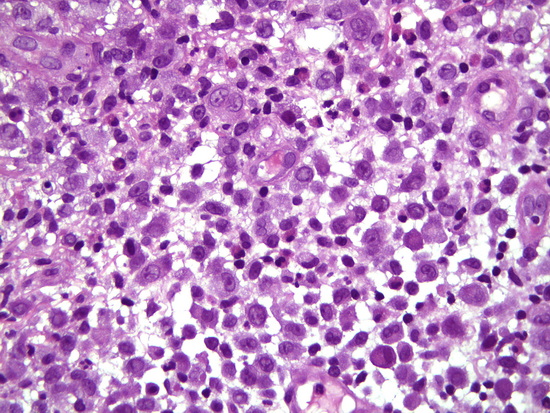
Рис 8 - увеличение х400, гематоксилин-эозин опухолевые клетки с овальными и удлиненными ядрами, складчатой формы с зубчатой поверхностью и нечеткими мелкими ядрышками, цитоплазма бледно-эозинофильная
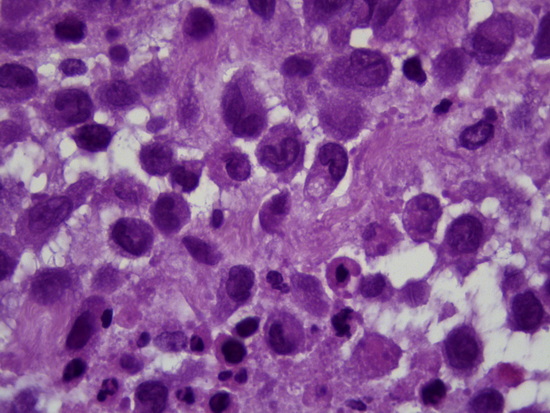
Рис 9 - увеличение х1000, гематоксилин-эозин опухолевые клетки, в ядрах обнаруживается продольная бороздка (ядра типа кофейного зерна)
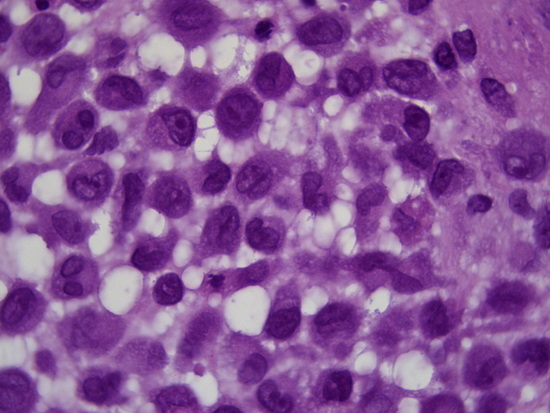
Рис 10 - увеличение х1000, гематоксилин-эозин опухолевые клетки, в ядрах обнаруживается продольная бороздка (ядра типа кофейного зерна).
В доставленном материале определяются фрагменты частично некротизированной опухоли, покрытые кожей. Опухоль представлена клеточными элементами среднего размера с овальными или удлиненными ядрами и мелкими нечеткими ядрышками, в ядрах многих клеток обнаруживается продольная борозда «ядра типа кофейного зерна», цитоплазма слабоэозинофильная. Так же отмечаются эозинофильные лейкоциты и многоядерные гигантские клетки. Гистологическая картина соответствует гистиоцитозу из клеток Лангерганса. Диагноз был также подтвержден при проведении иммуногистохимического исследования.
Мальчик был направлен для дальнейшего лечения в специализированное отделение.
Обсуждение.
Гистиоцитоз из клеток Лангерганса (ГКЛ) — наиболее распространенное в детской популяции гистиоцитарное заболевание, характеризующееся аккумуляцией СD1a+/CD207+/S-100+-клеток в органах и тканях с образованием гранулематозных или литических очагов. Клиническая картина заболевания разнообразна и варьирует от монофокального поражения до тяжелого жизнеугрожающего мультисистемного процесса. Ранее разные формы ГКЛ рассматривали как отдельные нозологии: гистиоцитоз Х, эозинофильная гранулема, болезнь Леттерера – Сиве, болезнь Хэнда – Шуллера – Кристиана. В 1985 г. было создано
Международное общество по изучению гистиоцитарных заболеваний — Histiocyte Society. Одним из первых результатов работы общества было объединение всех вышеуказанных заболеваний под единым названием «Гистиоцитоз из клеток Лангерганса» на основании исследований французского патолога C. Nezelof, обнаружившего сходство выявляемых при ГКЛ патологических клеток с внутриэпидермальными макрофагами — клетками Лангерганса эпидермиса.
Эпидемиология
Ежегодно выявляется от 0,5 до 10 случаев на 1 млн населения. Преимущественно болеют дети в возрасте до 15 лет, значительная доля пациентов — дети 1-го года жизни. Средний возраст детей на момент установления диагноза примерно 3 года. Соотношение мальчиков и девочек 2:1.
ГКЛ может возникать в любом возрасте, однако наиболее часто заболевание выявляется в возрасте от 0 до 4 лет, в последующем заболеваемость снижается. Ранний дебют ГКЛ (в возрасте до 2 лет) ассоциирован с высоким риском развития мультисистемных форм ГКЛ, тогда как моносистемные и монофокальные формы ГКЛ чаще выявляются у детей в возрасте старше 5 лет. Мультисистемная форма заболевания протекает с поражением «органов риска» (печени, селезенки) и недостаточностью гемопоэза и без лечения быстро прогрессирует с развитием полиорганной недостаточности и гибелью ребенка. Популяционная частота ГКЛ, по разным данным, составляет от 2 до 9 случаев на 1 млн детей в год. Наиболее часто ГКЛ встречается среди латиноамериканцев, реже —среди афроамериканцев. Факторы риска развития ГКЛ в настоящее время не определены. Исключение —высокий риск изолированного поражения легких у курильщика. У больных, перенесших ГКЛ, в дальнейшем отмечается более высокая, чем в среднем по популяции, частота развития злокачественных заболеваний, которая достигает среди взрослых уровня 32%. При этом наиболее часто диагностируются различные формы лейкозов.
Патогенез
Патогенез ГКЛ длительное время являлся предметом острых дискуссий. Анализ клеточного состава гистиоцитарных очагов выявил присутствие в них большого числа Т-лимфоцитов, эозинофилов и макрофагов. Лимфоциты, находящиеся внутри патологического очага, экспрессируют большое количество костимулирующих молекул и цитокинов, вызывая локальный «цитокиновый шторм», способствующий повреждению окружающих тканей. Была показана способность клеток ГКЛ выделять плейотропный белок остеопонтин, стимулирующий созревание Т-хелперов. Кроме того, остеопонтин обладает свойством активировать остеокласты, которые, в свою очередь, выделяют большое количество цитокинов, что также приводит к повреждению тканей, окружающих очаг специфического воспаления, и дальнейшему повреждению органов и систем органов, отчасти объясняя клиническую картину, наблюдаемую при ГКЛ.
Дальнейшее изучение генетической природы ГКЛ привело к выявлению новых патогенных вариантов в генах, кодирующих ключевые киназы MAPK-сигнального пути. Так, в результате анализа 40 BRAF-негативных образцов, полученных от больных с ГКЛ, в 28% случаев были обнаружены патогенные варианты в гене MAP2K. Позднее при ГКЛ были описаны патогенные варианты в гене ARAF, а также в генах NRAS, KRAS, HRAS, кодирующих одноименные киназы семейства RAS (rat sarcoma).
Модель нарушенной миелоидной дифференцировки
Для изучения происхождения ГКЛ из ранних миелоидных предшественников как молекулярные метки были использованы патогенные варианты гена BRAF. Анализ образцов крови и биоптатов костного мозга больных с мультисистемными формами ГКЛ с вовлечением «органов риска» показал наличие клеток с BRAF V600E патогенным вариантом как среди клеток миелоидной популяции периферической крови, так и в популяции СD34+-стволовых клеток костного мозга. Также было показано, что у больных с моносистемными формами заболевания в периферической крови мутантная популяция BRAF-позитивных клеток отсутствует, а у больных с мультисистемными формами с поражением «органов риска» выявляется чаще, чем без их поражения. В этой связи был сделан вывод, что тяжесть заболевания напрямую связана с онтогенетическим уровнем клетки-предшественницы, в которой происходит мутация. Патогенные варианты, возникшие в ранних костномозговых миелоидных предшественниках с высокой пролиферативной способностью и мультипотентным по тенциалом, приводят к крайне агрессивным мультисистемным формам ГКЛ с поражением «органов риска». Патогенные варианты, возникшие на уровне тканевого предшественника или резидентной дендритной клетки, приводят к мультисистемным формам ГКЛ без поражения «органов риска» или моносистемным формам заболевания.
Представления о механизмах нейродегенеративного поражения ЦНС при ГКЛ также изменились после открытия патогномоничных для заболевания патогенных вариантов в генах MAPK-сигнального пути. Исследования E. Mass и соавт. на мышиных моделях показали, что эритромиелоидные предшественники c патогенными вариантами в гене BRAF V600E, берущие начало в желточном мешке, могут заселять головной мозг BRAF V600E-позитивными клетками микроглии, что приводит к развитию прогрессирующей нейродегенерации у взрослых особей. Таким образом, соматические мутации, возникшие на уровне желточного мешка, потенциально могут приводить к развитию специфической нейродегенерации у больных с ГКЛ. Исследования биоптатов головного мозга больных со специфической нейродегенерацией при ГКЛ показали обширную инфильтрацию BRAF V600E-позитивными клетками с моноцитарным фенотипом, что потенциально подтверждает вышеописанную модель.
Клиническая картина
Основываясь на количестве вовлеченных в патологический процесс систем органов, выделяют моносистемную и мультисистемную формы ГКЛ. Среди моносистемных форм с поражением скелета выделяют монофокальные (1 очаг) и мультифокальные (2 и более очага) варианты заболевания. Наиболее тяжелое течение ГКЛ отмечается у больных с нарушением гемопоэза, поражением печени и селезенки.
Поражение костей скелета при ГКЛ возникает у 70–80% больных и протекает с выраженным болевым синдромом и патологическими переломами, приводящими к инвалидизации больного. При этом монофокальные формы заболевания встречаются чаще, чем мультифокальные. Патологические очаги при ГКЛ могут локализоваться в любых костях, однако более чем в 50% случаев происходит поражение плоских костей (череп, ребра, таз). Рентгенологическим признаком поражения костей при ГКЛ является литический очаг. Он представляет собой фокус снижения рентгеновской плотности кости, обычно округлой формы, с размытыми контурами.
Литические очаги при ГКЛ чаще всего локализованы в костях черепа. При этом поражение свода черепа встречается чаще, чем поражение его основания, с преимущественной локализацией литических очагов в париетальной и фронтальных областях.
В указанных случаях клиническая и радиологическая картина схожа с таковой при саркоме Юинга. Для дифференциальной диагностики необходима биопсия патологического очага с последующим иммуногистохимическим исследованием. Диагноз ГКЛ устанавливается при обнаружении CD1a-, CD207-, S100-позитивного клеточного материала.
Поражение кожи при ГКЛ происходит у 30–60% больных. Морфологическая картина кожного поражения разнообразна — это и покрытые коркой или чешуйчатые узелки, ограниченные участки некроза, папулы, а также сосудистые опухолеподобные образования. Распространенность кожных элементов также варьирует. Поражения могут затрагивать отдельный участок или вовлекать обширные области: кожу волосистой части головы, лица, области естественных складок на туловище, область ягодиц.
Наиболее часто кожные элементы локализуются на коже туловища. Изолированные кожные поражения при ГКЛ встречаются крайне редко, они характерны для младенцев мужского пола и ассоциированы с относительно благоприятным прогнозом. Особая форма врожденного кожного ГКЛ носит название болезни Хашимото – Притцкера и характеризуется наличием на коже саморазрешающихся красно-коричневых узелков. Поражение кожи у детей в возрасте 18 мес и старше, как правило, свидетельствует о мультисистемной форме заболевания. При ГКЛ описано также и поражение ногтей. Связь данного клинического феномена с прогнозом болезни не установлена. Однако у 50% процентов больных поражение ногтей ассоциировано с патологическим процессом в легких.
Поражение кишечника различных отделов или всего кишечника при ГКЛ происходит редко. Клиническая картина поражения кишечника включает в себя абдоминальный болевой синдром, тошноту, рвоту, диарею, в том числе с геморрагическим компонентом. Длительно текущее поражение кишечника может приводить к формированию синдрома мальабсорбции и/или белоктеряющей энтеропатии и осложняться в некоторых случаях перфорацией кишечника. При эндоскопическом исследовании могут быть обнаружены поверхностные эрозии и геморрагические изъязвления.
Поражение легких. Изолированное поражение легких у детей с ГКЛ встречается крайне редко (частота <1%). Клиническая картина характеризуется непродуктивным кашлем, возникновением одышки, в некоторых случаях присоединением неспецифического болевого синдрома, появлением цианоза, общей слабости, потерей массы тела. Приблизительно в 10% случаев у таких больных развивается пневмоторакс. Среди взрослых больных с ГКЛ изолированное поражение легких встречается в 24% случаев. Методом выбора при диагностике является компьютерная томография. Рентгенологические признаки поражения легких при ГКЛ схожи у взрослых и детей и характеризуются формированием нодулярных или ретикулонодулярных инфильтратов с последующей трансформацией в буллезнокистозные изменения. Как правило, поражения симметричны, с локализацией преимущественно в верхних и средних долях легкого. При длительном течении заболевания наблюдается развитие фиброза, что в некоторых случаях требует трансплантации легких.
Недостаточность гемопоэза. В соответствии с критериями Histiocyte Society, наличие цитопении по двум и более росткам кроветворения расценивается как недостаточность гемопоэза и ассоциировано с неблагоприятным прогнозом. Однако природа нарушения гемопоэза до конца не определена. Немногочисленные публикации, посвященные морфологии костного мозга при ГКЛ, основаны на рутинной микроскопии и не позволяют выявить корреляцию между гистологической картиной костного мозга и цитопенией. Исследование M. Minkov и соавт. показало, что присутствие в образцах костного мозга CD1a+-клеток, имеющих дендритную морфологию, является патогномоничными признаком нарушения гемопоэза при ГКЛ. При этом во всех исследованных образцах было обнаружено лишь небольшое количество CD1a+-клеток (единичные клетки либо скопление небольших клеточных кластеров). Таким образом, было показано, что цитопения при ГКЛ возникает не в результате замещения нормального гемопоэза патологическими клетками (механизм, характерный для лейкозов), а в результате иных, пока не установленных, причин. Тем не менее, согласно современным рекомендациям, поражение костного мозга при ГКЛ диагностируется только в случае выявления в биоптате CD1a+-клеток. В тех случаях, когда выявляются только лабораторные критерии цитопении, говорят о недостаточности гемопоэза без вовлечения костного мозга.
Поражение печени наряду с поражением других «органов риска» ассоциировано с неблагоприятным прогнозом. Клинически поражение печени может проявляться гепатомегалией, желтухой и асцитом, лабораторно — повышением активности трансаминаз, гипоальбуминемией, дефицитом факторов свертывания крови. Также при ГКЛ возможно поражение желчных протоков и, как следствие, развитие холестаза, склерозирующего холангита и билиарного цирроза. До 75% больных с ГКЛ со склерозирующим холангитом не отвечают на проводимую химиотерапию. Единственным возможным методом лечения таких пациентов является проведение трансплантации печени.
Поражение селезенки при ГКЛ может носить как первичный, так и вторичный характер и развиваться в результате портальной гипертензии. Как правило, поражение селезенки является частью мультисистемного процесса. Изолированное поражение селезенки при ГКЛ встречается крайне редко и описано в единичных клинических случаях. Селезенка является «органом риска», ее поражение связано с плохим прогнозом
Поражение лимфатических узлов чаще обнаруживается при мультисистемных формах ГКЛ. Как правило, наблюдается вовлечение шейных и грудных групп. Клинически поражение характеризуется увеличением размеров лимфатических узлов, локальным отеком, болевым синдромом.
Поражение ЦНС при ГКЛ, по разным данным, возникает в 3–57% случаев. Основным методом диагностики поражения ЦНС при ГКЛ является магнитно-резонансная томография (МРТ). Выделяют две основные клинические формы поражения ЦНС — туморозную форму и специфическую нейродегенерацию. В некоторых случаях наблюдается одновременное присутствие изменений на МРТ, характерных как для первой, так и для второй формы заболевания Наиболее частой локализацией туморозной формы являются мозговые оболочки, сосудистые сплетения и паренхима головного мозга. В зависимости от локализации патологического процесса проявления заболевания могут включать признаки нейроэндокринных нарушений, фокальные судороги, повышенное внутричерепное давление. Клиническая картина поражения передней доли гипофиза ассоциирована с явлениями центрального несахарного диабета, а также задержкой роста, задержкой / преждевременным половым созреванием, гипотиреозом, гипогонадизмом, гипокортицизмом или пангипопитуитаризмом. При МРТ очаги гранулематозного поражения имеют гипо-изоинтенсивный характер в Т1- и Т2-взвешенных режимах, усиливающийся после применения контраста. Дифференциальную диагностику обычно проводят с другими гистиоцитарными заболеваниями: болезнью Розаи – Дорфмана, ювенильной ксантогранулемой, а также с краниофарингиомами, саркоидозом и метастазами других опухолей.
Лечение
Попытки систематизировать подходы к терапии больных ГКЛ начались в начале 1980-х гг. В частности, в двух исследованиях, DAL HX-83 и AIEOP-CNR-H.X ’8, изучали эффективность различных режимов химиотерапии у больных с мультисистемными формами ГКЛ. Начиная с 1991 г. под эгидой Histiocyte Society были проведены глобальные многоцентровые исследования протоколов LCH I–IV, результаты которых заложили современные основы терапии больных с различными формами ГКЛ. В исследования LCH-I и LCH-II были включены только пациенты с мультисистемными формами заболевания. В исследовании LCH-III кроме пациентов с мультисистемными формами с поражением «органов риска» вошли пациенты с моносистемными формами заболевания. Схема лечения, примененная в протоколе LCH-III, включающая винбластин в сочетании с преднизолоном длительностью 12 мес, стала стандартом первой линии терапии для больных с мультисистемными формами ГКЛ.
После обнаружения патогенных вариантов генов MAPK-сигнального пути, ассоциированных с развитием ГКЛ, в лечении гистиоцитозов стала активно использоваться таргетная терапия, применяемая у больных при других онкологических заболеваниях со сходными вариантами генов (BRAF- и MEK-ингибиторы). Первое описание использования BRAF-ингибиторов при мультисистемной форме ГКЛ у младенца свидетельствовало о быстром ответе на проводимую терапию в виде разрешения всех клинических проявлений заболевания в течение 2 нед от начала лечения. В настоящее время активно ведутся исследования применения BRAF-ингибиторов в качестве монотерапии и в комбинации с химиопрепаратами у педиатрических больных с ГКЛ. Опыт применения MEK-ингибиторов при ГКЛ ограничен. Так, в исследовании E. Diamond и соавт. включавшем 18 взрослых больных с различными заболеваниями из группы гистиоцитозов, эффективность MEK-ингибиторов (сокращение размеров ранее выявляемых литических очагов) была зафиксирована в 89% случаев. Исследование эффективности MEK-ингибиторов у детей показало сокращение размеров ранее выявляемых литических очагов в ответ на проводимую терапию в 62,5% случаев. Описаны единичные клинические случаи успешного применения комбинации BRAF- и MEK-ингибиторов у больных с гистиоцитарными заболеваниями.
Заключение
На протяжении многих лет природа и патогенез ГКЛ оставались не изучены и были предметом научных дискуссий. Успехи последних лет в понимании молекулярных механизмов ГКЛ привели к изменению стандартных терапевтических подходов. Применение таргетной терапии позволяет достигать ответа на лечение у всех групп больных с ГКЛ, однако отмена терапии во многих случаях приводит к быстрой прогрессии заболевания.
Таким образом, на сегодняшний день до конца не определены оптимальные режимы таргетной терапии, комбинации препаратов. Отсутствие длительных наблюдений за реконвалесцентами ГКЛ после окончания таргетной терапии также не позволяет с уверенностью говорить об эффективности таргетной терапии для профилактики развития «перманентных осложнений» ГКЛ.
Родителям необходимо обращать внимание на состояние кожи, особенно на голове. При выявлении объёмных образований следует сразу обращаться к специалистам. Только совместная работа детских хирургов, оториноларингологов, рентгенологов, патологоанатомов в данном случае помогла установить правильный диагноз и направить мальчика в специализированное отделение для проведения системной терапии.
Литература
Бурцев Е.А., Бронин Г.О. Вопросы современной педиатрии. 2023;22(1):13–22. doi: https://doi.org/10.15690/vsp.v22i1.2520
Данные об авторах.
- Стоногин Сергей Васильевич – врач-хирург хирургического отделения ГБУЗ ДГКБ имени З.А. Башляевой ДЗМ, кандидат медицинских наук. E-mail: svas70@mail.ru Адрес: 125480 Москва, улица Героев Панфиловцев, дом 28, отделение хирургии. https://orcid.org/orcid 0000-0003-3531-5849
- Царева Анна Игоревна – заведующая отделением оториноларингологии ГБУЗ ДГКБ имени З.А. Башляевой ДЗМ. E-mail: gutvan87@mail.ru Адрес: 125480 Москва, улица Героев Панфиловцев, дом 28, отделение оториноларингологии.
- Горюнов Алексей Константинович – врач-рентгенолог отделения лучевой диагностики ГБУЗ ДГКБ имени З.А. Башляевой ДЗМ. E-mail: goba79@mail.ru Адрес: 125480 Москва, улица Героев Панфиловцев, дом 28, отделение лучевой диагностики
- Таширова Екатерина Александровна – врач-патологоанатом отделения патологической анатомии ГБУЗ ДГКБ имени З.А. Башляевой ДЗМ. E-mail: tashirova.katya@mail.ru Адрес: 125480 Москва, улица Героев Панфиловцев, дом 28, отделение патологической анатомии. https://orcid.org/0000-0002-5303-4349
Diagnosis of Langerhans Cell Histiocytosis in a Three-Year-Old Boy S.V. Stonogin, A.I. Tsareva, A.K. Goryunov, E.A. Tashirova DGB Children's Hospital named after Z.A. Bashlyaeva, Moscow.
A three-year-old boy was admitted to the surgical department on an emergency basis. For a month, he had been experiencing pain, a mass in the left postauricular region, and an increase in the size of the mass over seven days.
In the left postauricular area, a subcutaneous mass approximately 2.5 cm in diameter, of soft-elastic consistency, moderately painful upon palpation, was detected. There was swelling and erythema up to 2 cm in diameter. The fluctuation symptom was positive.
The boy underwent emergency surgery. An incision of 0.5 cm was made over the fluctuation site. A scanty purulent discharge was observed. A white tumor-like mass (atheroma?) was identified. Bacterial culture of the wound exudate showed no growth of microflora.
Fig. 1 – View of the wound in the left postauricular region during dressing.
Fig. 1a – View of the wound in the left postauricular region during dressing.
Otorhinolaryngological status. The shape of the external nose and the projection of the paranasal sinuses were unchanged, and palpation was painless. The mucosa was pink and non-edematous. There was scanty mucous discharge in the nasal passages. Nasal breathing was satisfactory. The right postauricular region was unchanged. In the left postauricular region, there was a wound with an elastic mass protruding from it, covered with fibrin, measuring 2 × 2 cm. The skin over the mastoid process was erythematous. The auditory canals were wide and clear. The tympanic membranes were dull.
Oropharynx: The pharynx was symmetrical, tonsils were grade 2 and hyperemic. The posterior pharyngeal wall showed no abnormalities.
Given the uncharacteristic appearance of the wound for an abscess, the lack of response to antibiotic therapy and antiseptic dressings, and the persistence of pain upon palpation, a spiral computed tomography (CT) scan of the head was performed.
A series of tomograms with a slice thickness of 1 mm was obtained, providing images of the brain. The midline structures were not shifted. The lateral ventricles were symmetrical and not dilated; the third ventricle was not dilated; the fourth was not deformed.
The basal cisterns were not dilated or deformed. The perimesencephalic cisterns were symmetrical and not dilated. The cerebral sulci: lateral sulci were symmetrical and not dilated; the interhemispheric fissure was not dilated. Convexital sulci were unchanged. Differentiation between gray and white matter was preserved. No additional masses or foci of abnormal density were detected. The subarachnoid spaces were not dilated. The cranial vault bones were unchanged. The paranasal sinuses and ethmoidal cells, as well as the mastoid cells on the right and the middle ear cavities on both sides, were pneumatized. The structures of the pyramids of the temporal bones were preserved and symmetrical.
In the projection of the left mastoid process and postauricular region, an area of bone destruction was observed with the loss of the outer cortical plate, walls of the mastoid cells, and the inner cortical plate in the projection of the sigmoid sinus groove. Within the lytic bone area, a soft tissue structure of heterogeneous density, averaging about 45 HU, was noted.
CT findings indicated a mass in the projection of the left mastoid process.
Fig. 2 – Computed tomography of the head. 3D reconstruction.
Fig. 3 – Computed tomography of the head. 3D reconstruction.
Fig. 4 – Computed tomography of the head.
Following the CT scan, a biopsy of the mass was performed, and the entire mass was excised and sent for histological examination.
Fig. 5 – Macroscopic specimen of the removed mass.
Fig. 6 – Magnification ×40, hematoxylin-eosin staining of the skin, tumor in the dermis.
Fig. 7 – Magnification ×100, hematoxylin-eosin staining of the skin, tumor in the dermis.
Fig. 8 – Magnification ×400, hematoxylin-eosin staining of tumor cells with oval and elongated nuclei, folded shape with a serrated surface and small indistinct nucleoli, cytoplasm pale eosinophilic.
Fig. 9 – Magnification ×1000, hematoxylin-eosin staining, tumor cells with nuclei exhibiting a longitudinal groove (coffee bean nuclei).
Fig. 10 – Magnification ×1000, hematoxylin-eosin staining, tumor cells with nuclei exhibiting a longitudinal groove (coffee bean nuclei).
The delivered material contained fragments of partially necrotic tumor covered with skin. The tumor was composed of medium-sized cells with oval or elongated nuclei and small indistinct nucleoli. Many nuclei exhibited a longitudinal groove, "coffee bean nuclei." The cytoplasm was weakly eosinophilic. Eosinophilic leukocytes and multinucleated giant cells were also noted. The histological picture corresponded to Langerhans cell histiocytosis. The diagnosis was confirmed by immunohistochemical analysis.
The boy was referred for further treatment to a specialized department.
Discussion.
Langerhans cell histiocytosis (LCH) is the most common histiocytic disorder in children, characterized by the accumulation of CD1a+/CD207+/S-100+ cells in organs and tissues, forming granulomatous or lytic lesions. The clinical presentation of LCH is diverse, ranging from monofocal involvement to severe, life-threatening multisystem disease. Previously, different forms of LCH were considered separate nosological entities: Histiocytosis X, eosinophilic granuloma, Letterer-Siwe disease, and Hand-Schüller-Christian disease. In 1985, the International Histiocyte Society was established.
One of the first outcomes of the society's work was the unification of all the above-mentioned diseases under the single term "Langerhans cell histiocytosis" based on studies by the French pathologist C. Nezelof, who identified similarities between LCH pathological cells and intraepidermal macrophages—Langerhans cells of the epidermis.
Epidemiology
The incidence of LCH is between 0.5 and 10 cases per million population annually. It predominantly affects children under 15 years of age, with a significant proportion of patients being infants. The median age at diagnosis is approximately three years. The male-to-female ratio is 2:1.
LCH can occur at any age but is most frequently diagnosed between 0 and 4 years, with incidence declining thereafter. Early-onset LCH (before the age of two) is associated with a high risk of multisystem involvement, whereas monosystemic and monofocal forms are more commonly diagnosed in children over five years old. The multisystem form progresses rapidly without treatment, leading to multiorgan failure and death.
The population frequency of LCH varies from 2 to 9 cases per million children annually. It is most common among Latin Americans and less frequent among African Americans. Risk factors for LCH remain unidentified, except for a higher risk of isolated lung involvement among smokers. LCH survivors have an increased risk of malignancies later in life, reaching up to 32% among adults, with leukemia being the most frequently diagnosed malignancy.
[The document continues with a discussion on pathogenesis, genetic findings, and clinical presentation.]